Intravascular large B-cell lymphoma (IVLBCL) is a rare form of extranodal diffuse large B-cell lymphoma characterized by the proliferation of lymphoma cells in the lumen of small blood vessels. Clinical presentation varies among reported cases and diagnosis can be challenging for both clinicians and pathologists. We report a case of a 64-year-old female with a history of prolonged fever. Diagnosis was suspected clinically due to thrombocytopenia and elevated serum lactate dehydrogenase after exhausting work-up for an infectious etiology. IVLBCL was established on post-mortem examination upon finding infiltration of CD20-positive neoplastic cells in several organs in the absence of lymph node involvement.
Key words: lymphoma, large B cell, intravascular, fever of unknown origin
Intravascular large B-cell lymphoma (IVLBCL) is a rare type of non-Hodgkin’s lymphoma found in adults. Since its discovery in the 1960’s, most of the available information regarding this disease entity has been collected through individual case reports, with an estimated incidence of less than one person per million. Few reports cover large series of cases. A study of 38 Western patients diagnosed with intravascular lymphoma was made in 2004 in relation to clinical presentation and natural history.[1] The largest study to date reported a series of 96 patients diagnosed with IVLBCL in Japan, addressing clinical profiles, outcomes and prognostic factors.[2]
IVLBCL is characterized by the selective growth of neoplastic cells within the blood vessel lumina.[3] It has been dubbed “the oncologist’s great imitator” due to its protean manifestation. The disease manifests as fever of unknown origin and is frequently diagnosed post-mortem due to its aggressive clinical course.[4] Neoplastic cells are not usually seen in the peripheral blood smear, but laboratory findings of anemia, elevated serum lactate dehydrogenase, thrombocytopenia and leukopenia should raise a suspicion for IVLBCL.[5] Increased awareness of this disease entity, for both clinicians and pathologists, may clinch the diagnosis in the future.
This is a case of a 64-year old female who was admitted due to a three-week history of intermittent fever. She had no associated symptoms such as cough or colds. The past medical, personal and social history were unremarkable. Physical examination revealed palpable cervical lymph nodes and lower gastrointestinal bleeding secondary to hemorrhoids; the rest were unremarkable. Several diagnostic work-ups were done to determine the etiology of the fever. Complete blood counts show anemia (8.8 g/dL) and thrombocytopenia (71-137 x 10^9/L). She had normal levels of white blood cells with predominance of segmenters. Blood culture was negative. CT scan of the chest reveal non-specific pulmonary nodules in the right lung and cardiomegaly. To rule out a malignant etiology of gastrointestinal bleeding, abdominal CT scan was also done which showed dilated and tortuous blood vessels on both liver lobes and esophageal varices, suggestive of portal hypertension. These findings, however, could not explain the cause of fever. Gastroscopy and colonoscopy findings were non-contributory. During the hospital stay, she developed cough associated with difficulty of breathing. Chest radiograph showed streaky hazy infiltrates in both bases for which she was managed as a case of hospital-acquired pneumonia and started on antibiotics. Work-up for autoimmune disorders (ANA, C3 and RF) were negative. At this time, a hematolymphoid malignancy was being considered. Bone marrow biopsy findings showed a variably cellular marrow (70-90%) with mild panhyperplasia and slight lymphocytosis. No definite lymphoid aggregates or granulomatous infiltrates are seen. A correlation with clinical findings was recommended. Serum lactate dehydrogenase was requested and was found to be elevated at 3349 U/l. On her 3rd hospital week, the patient had episodes of waxing and waning of sensorium associated with a rigid neck. Cranial MRI and CSF studies were done to rule out meningitis which all turned out to be unremarkable. With the patient’s history of prolonged fever without an infectious focus and a significantly elevated LDH (6000 U/l), consideration at that time was leaning towards intravascular lymphoma. Further work-up was done at the interim, but bone marrow biopsy results were still negative. The condition of the patient gradually deteriorated and she succumbed on the 24th hospital day.
Autopsy was performed 17 hours after the patient’s demise. On external examination, she had multiple flat, irregular, erythematous lesions distributed on her left shoulder, right forearm, abdomen and lower extremities (Figure 1). There were no palpable masses or lymphadenopathies. Cardiomegaly (430 grams; normal 179-385 grams) and splenomegaly (265 grams; normal 55-195 grams) was noted after weighing all the organs. The right and left ventricular walls were hypertrophic. Both lungs had boggy, subcrepitant parenchyma. The liver was not enlarged. Although smooth upon palpation, the parenchyma appeared to have small nodules which led us to assume the patient has micronodular cirrhosis which would explain her signs of portal hypertension. Macroscopic examination of the gastrointestinal tract showed esophageal varices and gastric ulcers and a colonic polyp. Infarcts were noted the base of the pons, left cerebellar peduncle and left internal capsule (Figure 2). The endocrine and genitourinary organs were unremarkable. Representative sections were taken and submitted for histopathological examination.
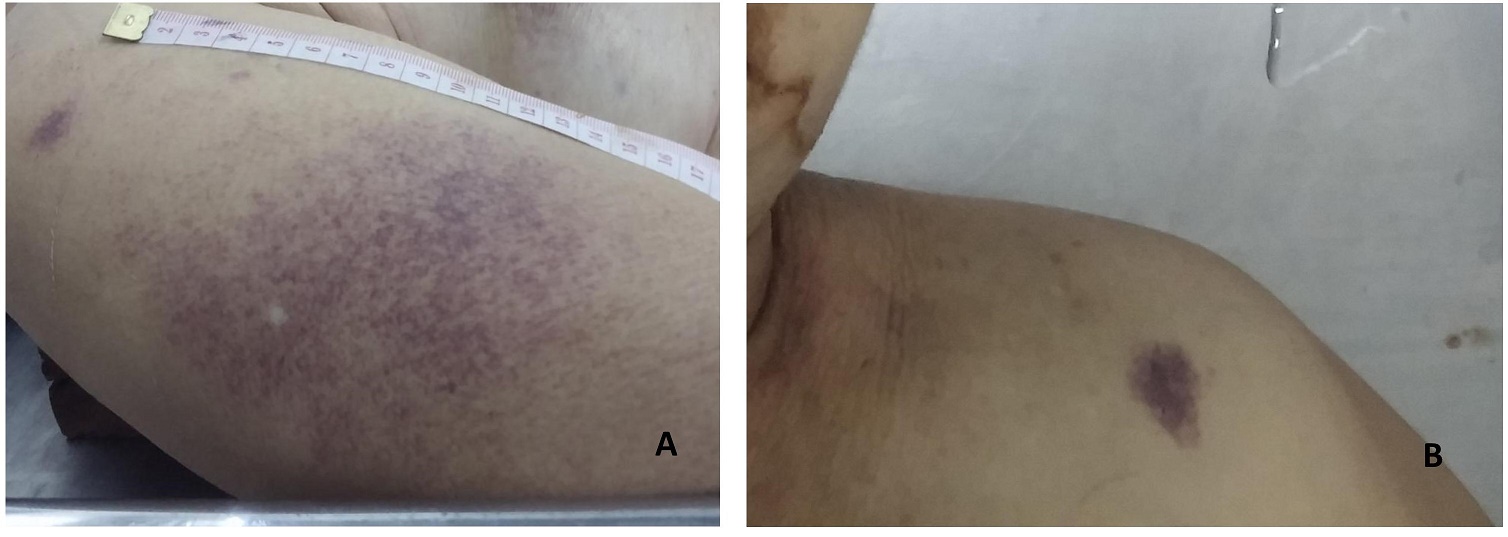
Figure 1. Skin lesions. (A) Flat, irregular, red to violaceous, rash-like lesion on the right upper arm; (B) Flat skin lesion on the left shoulder.
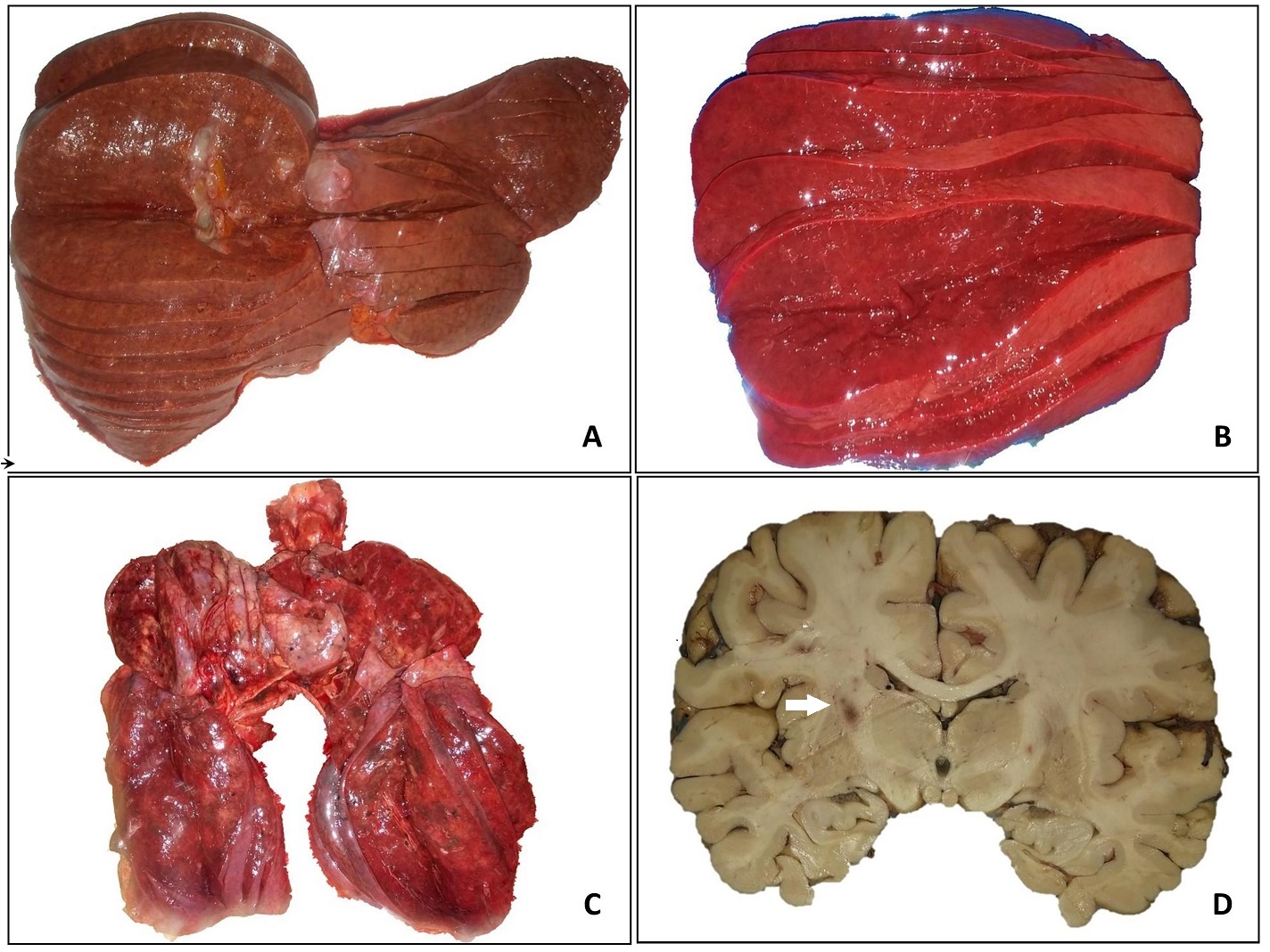
Figure 2. Gross appearance of the organs. Cut sections of the liver (A) and spleen (B). The right and left lungs (C) have red to brown, soft, boggy, sub-crepitant parenchyma; air spaces are not enlarged. Focal, dark brown, infarct-like lesions are noted on the brain parenchyma. The arrow points to the lesion seen in the internal capsule (D).
Microscopic examination reveal aggregates of atypical lymphoid cells in the small to medium-sized blood vessels and sinusoids of the following organs: lungs, liver, spleen, kidneys, uterus, cervix, brain, heart and thyroid. These cells have increased nuclear-to-cytoplasmic ratio, irregular nuclei, some with prominent nucleoli. Immunohistochemical staining revealed positivity for B-cell marker CD20, and negative for CD3, CD10 and CD30. Post-mortem bone marrow sample was noted to be hypercellular (70-80%) with trilineage maturation. Immunohistochemical staining with CD20, however, was negative. The findings of intravascular neoplastic proliferation of atypical lymphocytes in several organs, absence of lymph node involvement, and positivity for CD20 led us to the diagnosis of intravascular diffuse large B-cell lymphoma (Figure 3).
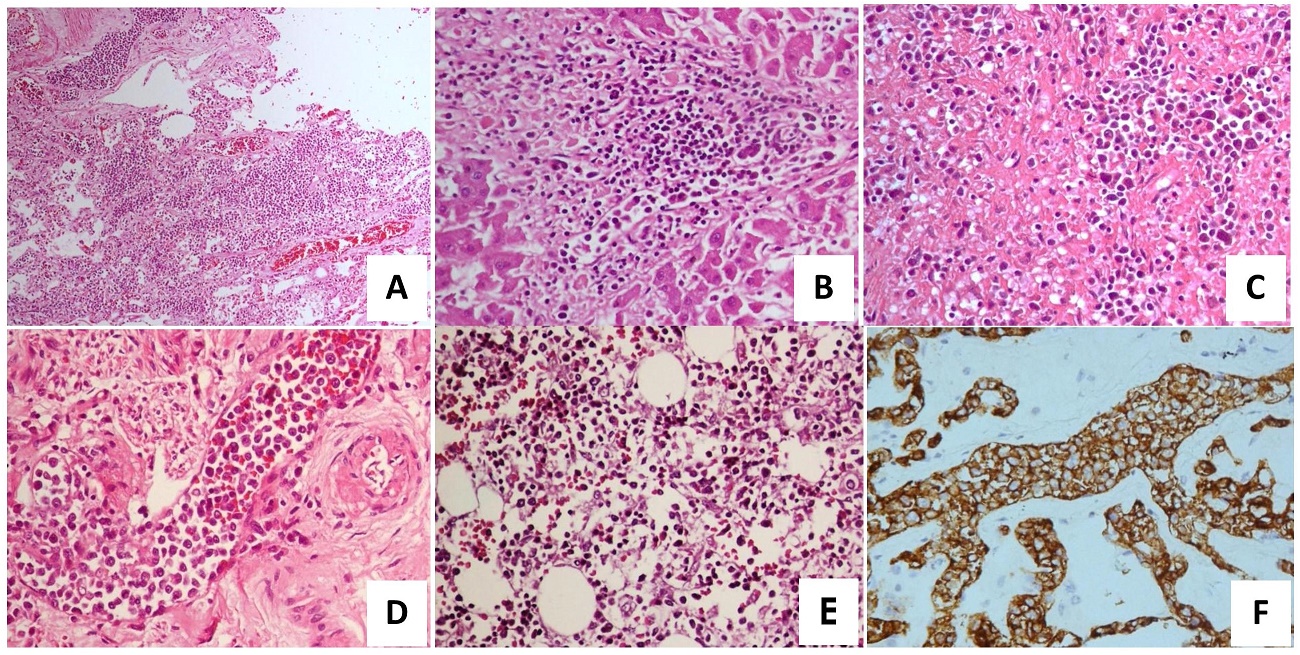
Figure 3. Intravascular large cell B-lymphoma. Proliferation of neoplastic lymphoid cells within the lumen of small to medium-sized blood vessels are noted in the following organs: (A) Lungs, H&E, 10x (B) Liver, H&E, 40x (C) Spleen, H&E, 40x (D) Uterus, H&E, 40x. (E) Hypercellular bone marrow with trilineage maturation. Immunohistochemistry with CD3, CD20 and CD30 were all negative. (F) Neoplastic cells found in the blood vessels of the lungs show strong, cytoplasmic reactivity to CD20, 40x. The same immunohistochemistry pattern was noted in the liver, spleen, kidneys, uterus, cervix, brain, heart and thyroid. .
Intravascular large B-cell lymphoma (IVLBCL) is a rare form of extranodal diffuse LBCL characterized by proliferation of lymphoma cells within the lumina of vessels, particularly capillaries, with exception of large arteries or veins.[3] It was previously called angiotropic large cell lymphoma. This entity is typically found in adults, with a median age of 67 years, and is found to have equal distribution among both sexes. It has an aggressive clinical course, usually widely disseminated and may present virtually on any organ. Lymph nodes are usually spared. Intravascular growth pattern has been hypothesized to be secondary to a defect in homing receptors on the neoplastic cells such as lack of CD29 and CD54 (ICAM-1) adhesion β-molecules.[4] Fever of unknown origin is a prominent sign and is seen in approximately 45% of cases.[5] Laboratory findings are not specific but should raise IVLBCL suspicion, and these include anemia, elevated serum lactate dehydrogenase, elevated erythrocyte sedimentation rate, thrombocytopenia, leukopenia and hypoalbuminemia. Majority of the studies have shown neoplastic cells to be absent in peripheral blood examination.[6] Western and Asian variants have been identified based on clinical presentation. Patients from Western countries display a high frequency of CNS and skin involvement, while those from Asian countries show hemophagocytic syndrome, bone marrow involvement, fever, hepatosplenomegaly and thrombocytopenia.[7] In a clinicopathological study of diagnosed IVLBCL cases in China, 8 out of 13 patients displayed bone marrow involvement, supported by immunohistochemistry. They concluded that a morphological “sinusoidal pattern” is essential to the diagnosis of IVLBCL in a bone marrow biopsy.[8] In our case, however, bone marrow biopsy results were negative.
Hepatic injury was clinically evident in this case. In the wards, she was noted to have elevated serum ammonia, SGPT, alkaline phosphatase and bilirubin levels. Esophageal varices and hemorrhoids support the diagnosis of portal hypertension on Doppler ultrasound. Consequently, the increased portal blood flow led to enlargement of the spleen. Another IVLBCL case with the same manifestation was reported in a 62-year old male,[9] but no thorough discussion was made regarding the portal hypertension. Microscopically, the liver of our patient was not cirrhotic. Histologic examination showed diffuse sinusoidal infiltration of the liver parenchyma. Lymphoma has been cited as a cause neoplastic occlusion of the portal vein.[10] Though IVLBCL is said to be restricted to the smallest of blood vessels, one case report mentioned the embolization of lymphoma cells in the hepatic portal vein, splenic vein and mesenteric vein.[11]
Cutaneous involvement varies in appearance and may present as painful indurated erythematous eruptions, poorly circumscribed violaceous plaques, to palpable purpura or small red palpable spots. Unfortunately for our case, a skin biopsy was not carried out on post-mortem examination. One study recommends performing skin biopsy irrespective of the presence or absence of skin lesions in patients who are suspected to have IVLBCL after they have concluded its high sensitivity compared to bone marrow biopsy.[12] Symptoms secondary to CNS involvement are common and varied and may range from sensory and motor deficits, altered conscious state, paresthesias to seizures.[13]
Quite unusual in this case is the presence of the neoplastic cells confined in the small blood vessels of the uterus and cervix. Although it has been frequently mentioned in different studies that IVLBCL can virtually involve any organ, the infiltration of the blood vessels in the gynecologic tract is rarely noted. At least 3 case reports describe the diagnosis of IVLBCL in the uterus, cervix and ovaries[14]-[16]—all of which noted the absence of a defined mass macroscopically, and confirming the diagnosis on histopathological examination.
Patients with IVLBCL have a poor prognosis, partly due to its protean manifestation and diagnostic delays. With its high overall mortality rate, more than half of the patients are diagnosed post-mortem.[17] It responds poorly to chemotherapy and neither clinical types nor clinical parameters can predict survival, apart from better prognosis for cases with disease limited to the skin.
Despite numerous efforts, a definitive diagnosis of B-cell lymphoma was not established before the patient’s demise. In an adult patient who presents with fever of unknown origin associated with anemia, elevated serum lactate dehydrogenase, thrombocytopenia and leukopenia, altered hepatic, renal or thyroid function tests, a differential diagnosis of IVLBCL should be considered. A bone marrow or skin biopsy may be helpful. However, should the case present as a clinical dilemma such that diagnosis cannot be rendered prior to the patient’s demise, postmortem examination is recommended to further increase the awareness for this disease entity and to better understand its pathogenesis.
[1] Ferreri A, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-83. PbMed DOI.
[2] Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathological study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-85. PubMed DOI.
[3] Nakamura S, Ponzoni M, Campo E. Intravascular large B-cell lymphoma. In S. Swerdlow, E. Campo, Harris NL, editors. WHO classification of tumours of haematopoeitic and lymphoid tissues, 4th ed. Lyon: IARC. 2008, pp.252-3.
[4] Ponzoni M, Arrigoni G, Gould VE, et al. Lack of CD29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol. 2000;31(2):220-6. PubMed.
[5] Holmes NE, Gordon CL, Lightfoot N, et al. Intravascular large B cell lymphoma: an elusive cause of pyrexia of unknown origin diagnosed postmortem. Clin Infect Dis. 2010; 51(9): e61-4. PubMed DOI.
[6] Zuckerman D, Seliem R, Hochberg E. Intravascular lymphoma: the oncologist's "great imitator". Oncologist. 2006;11(5):496-502. PubMed DOI.
[7] Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-73. PubMed DOI.
[8] Wang J, Ding W, Gao L, et al. High frequency of bone marrow involvement in intravascular large B-cell lymphoma: a clinicopathological study of 13 cases in China. Int J Surg Pathol. 2016;25(2):118-26. PubMed DOI.
[9] Fiegl M, Greil R, Pechlaner C, Krugmann J, Dirnhofer S. Intravascular large B cell lymphoma with a fulminant clinical course: a case report with definite diagnosis post mortem. Ann Oncol. 2002;13(9):1503-6. PubMed.
[10] Khanna R, Sarin S. Non-cirrhotic portal hypertension--diagnosis and management. J Hepatol. 2014;60(2):421-41. PubMed DOI.
[11] Yasuda, H, Ando J, Matsumoto T, et al. Intravascular large B cell lymphoma with hepatic portal vein, splenic vein and mesenteric vein tumour embolism. Histopathology. 2010;57(4):648-50. PubMed DOI.
[12] Matsue K, Asada N, Odawara J, et al. Random skin biopsy and bone marrow biopsy for diagnosis of intravascular large B cell lymphoma. Ann Hematol. 2011;90(4):417-21. PubMed DOI.
[13] Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-8. PubMed DOI.
[14] Sur M, Ross C, Moens F. Intravascular large B-cell lymphoma of the uterus: a diagnostic challenge. Int J Gynecol Pathol. 2005;24(2):201-3. PubMed.
[15] Shigematsu Y, Matsuura M, Tsuyama N, et al. Intravascular large B-cell lymphoma of the bilateral ovaries and uterus in an asymptomatic patient with a t(11;22)(q23;q11) constitutional translocation. Intern Med. 2016;55(21):3169-74. PubMed PubMed Central DOI.
[16] Fournier M, Hourseau M, Chis C, Luton D, Koskas M. Uterine presentation of an intravascular large B-cell lymphoma diagnosed by PET/CT and histology. Case Rep Clin Pathol. 2014;1(2):105-8. DOI.
[17] Wahie S, Dayala S, Husain A, Summerfield G, Hervey V, Langtry JA. Cutaneous features of intravascular lymphoma. Clin Exp Dermatol. 2011;36(3):288–91. PubMed DOI.