Dedifferentiated Chordomas are rare variants of a malignant bone tumor arising from remnants of the embryonal notochord. Most cases are reported from chordomas that have recurred after surgical resection and/or radiation. Dedifferentiated Chordomas have an overall poorer prognosis compared with conventional chordomas due to their highly aggressive behavior and high metastatic potential. We report a case of a Dedifferentiated Chordoma from the sacrum in a 53-year-old female with no known prior surgery or radiation treatment. The associated clinical and radiologic features are discussed along with a review of the reported cases in the English literature. The diagnostic pitfalls and approach for Chordomas as well as the current and developing treatment modalities are also reviewed.
Key words: chordoma, dedifferentiated chordoma, bone tumor, bone malignancy
Chordomas are low to intermediate grade, malignant tumors that arise from remnants of the embryonal notochord. These tumors are locally aggressive and have a propensity to recur and metastasize.[1] They are rare, with an incidence rate of less than 0.1 per 100,000 per year. According to most sources, chordomas account for approximately 5% of all malignant primary bone tumors.[2] Chordomas are most common in adults greater than 30 years of age, with a peak in incidence in the 6th decade. It is rare in patients less than 20 years old. Males are twice more commonly affected than females.[1]
There are three (3) recognized histologic variants: conventional, chondroid, and dedifferentiated. Another occasionally noted variant is the sarcomatous type. The axial spine is the most commonly-involved site. 60% of tumors are in the sacral spine, 25% occur in the spheno-occipital area, 10% are cervical, and 5% are thoracolumbar. Extra-axial sites are exceedingly rare. Surgery is considered the mainstay of treatment.[1]
The dedifferentiated variant of chordoma was first described by Meis et al., (1987).[3] It is characterized histologically as a tumor containing areas of conventional chordoma with a high-grade sarcomatous component, these areas being admixed and sharply demarcated.[2] There have been 16 reported cases in the English literature since 1970 that fulfill these specific criteria.[4] The dedifferentiated chordoma is discriminated from a sarcomatous chordoma wherein a transitional component between the conventional and sarcomatous elements exists in the latter. In terms of behavior and prognosis, however, there is no difference between the two.[4]
A 53-year-old Filipino female presented with occasional back pain two (2) years prior to surgery. The pain was managed with intake of pain relievers. The patient did not report urinary incontinence, stool retention, tenesmus, sciatic-like pain or paresis of the lower extremities. The patient’s personal and family medical histories were non-contributory. This continued until five (5) months prior to surgery when the back pain was noted to have worsened and was no longer relived by pain medications. Physical examination revealed a non-tender, movable mass on the left gluteal area. Abdominal ultrasound revealed a pelvic mass, initially considered to be an ovarian neoplasm. Exploratory laparotomy was done, revealing an unremarkable uterus and bilateral adnexa. Intraoperatively, a presacral mass was noted.
One (1) month prior to surgery, Magnetic Resonance Imaging (MRI) of the pelvic area was performed. A large, mixed intensity, lobulated, soft tissue presacral mass measuring 12.9 x 14.1 x 11.9 centimeters was noted. A chordoma was considered radiographically and clinically. The patient then underwent excision. Intraoperatively, the presacral mass was noted to have extended to the left gluteal area.
The primary mass measured 8 x 7.5 x 6 cm. The mass has cream-tan to tan, solid cut surfaces with foci of hemorrhage and necrosis. It is surrounded by a 0.2 cm-thick fibrous tissue. It is clearly-delineated from the surrounding muscle. Microscopic examination revealed the two (2) histologic components for the diagnosis of dedifferentiated chordoma. It consists of areas compatible with a conventional chordoma (Figures 1 and 2) and areas with sarcomatous differentiation (Figure 3). No transitional areas were seen between these components.
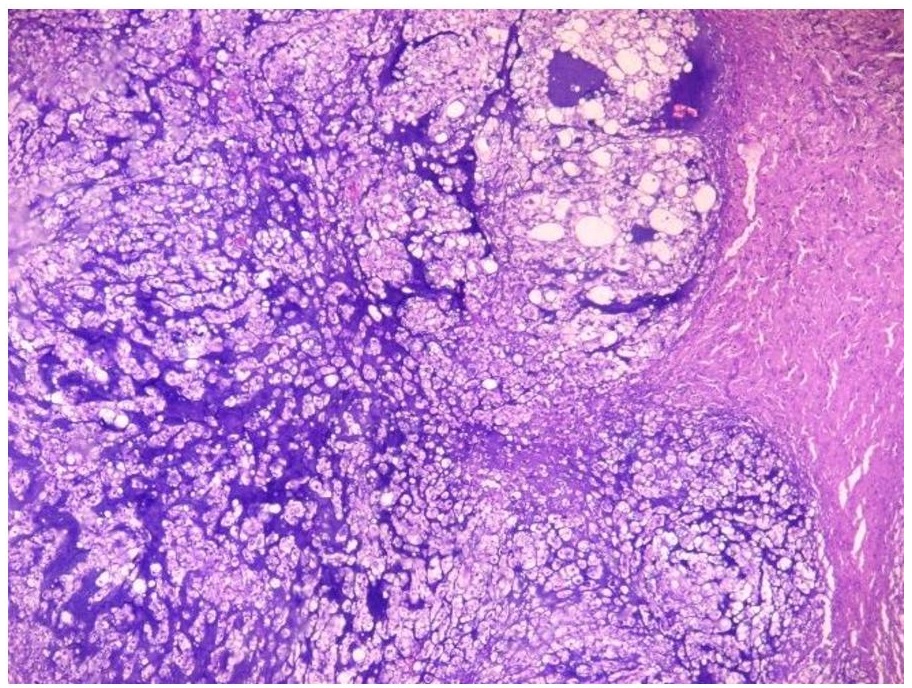
Figure 1. The tumor cells are infiltrative and are arranged in sheets, nests and chords. These areas are divided into lobules by fibrous septa (Hematoxylin and Eosin, 20x).
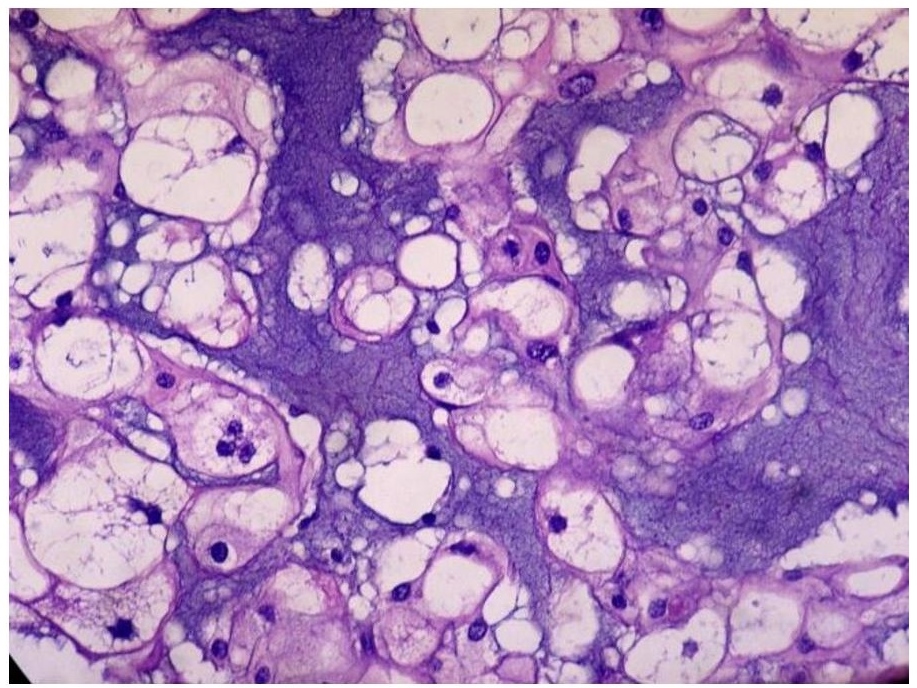
Figure 2. Cells with abundant pale, vacuolated/bubbly cytoplasm with small, hyperchromatic nuclei (physaliferous cells) seen alongside cartilage-like areas. There is mild to moderate nuclear atypia and rare mitoses (Hematoxylin and Eosin, 40x).
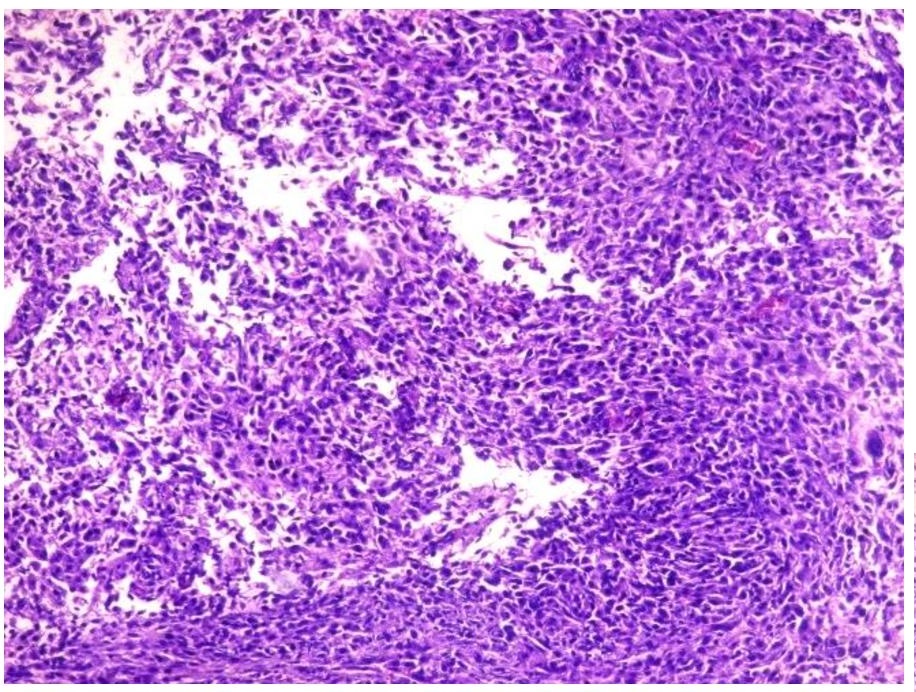
Figure 3. Dedifferentiated area with frank and pleomorphic, sarcomatous morphology (Hematoxylin and Eosin, 20x).
In addition, immunohistochemistry with Cytokeratin (CK), Epithelial Membrane Antigen (EMA) and S100 (Figures 4, 5 and 6) revealed moderate to strong staining in the tumor cells.
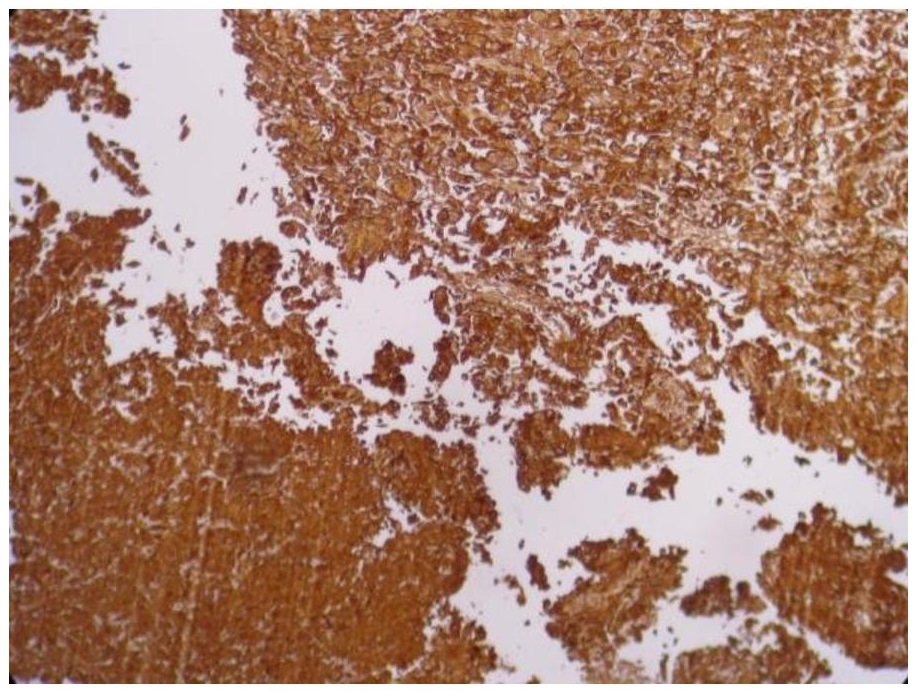
Figure 4. Strong cytoplasmic and membranous staining with Cytokeratin in the tumor cells, including the sarcomatous areas (Cytokeratin, 20x).
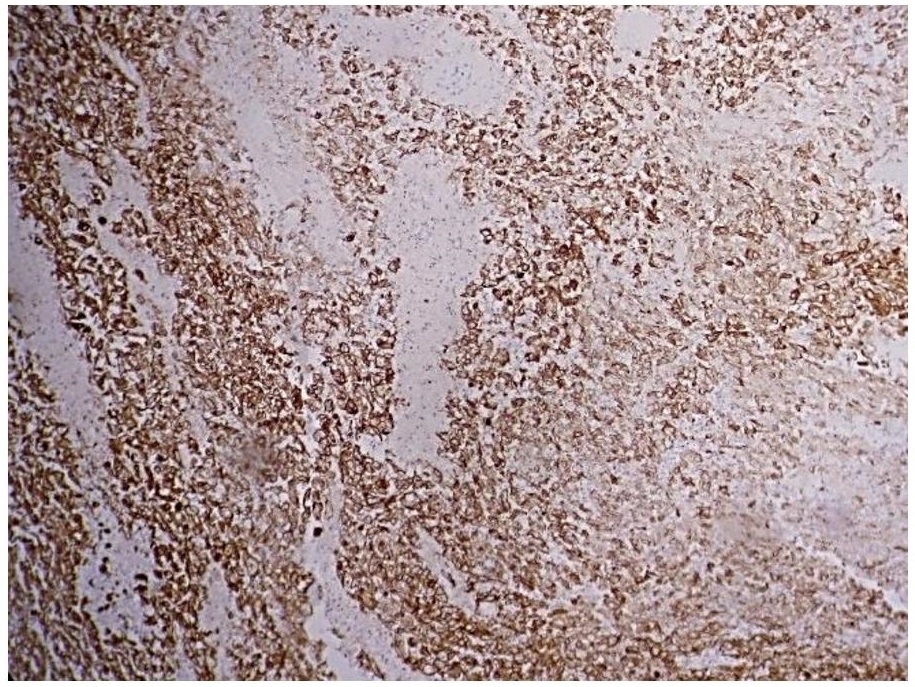
Figure 5. Moderate membranous staining with Epithelial Membrane Antigen (EMA, 20x).
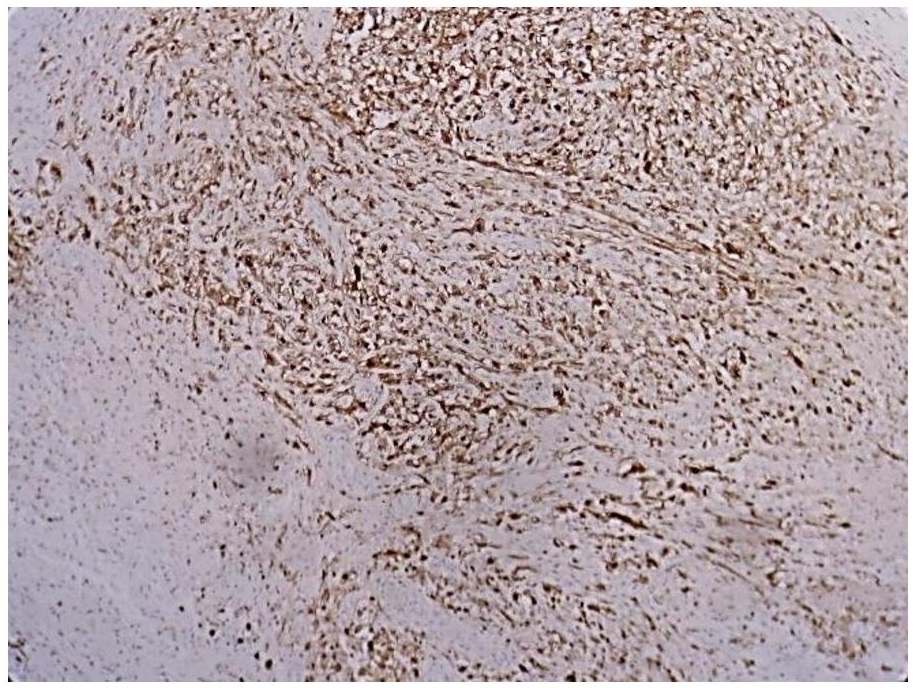
Figure 6. Moderate cytoplasmic and membranous staining with S100 (S100, 20x).
As of this writing, the patient has undergone radiotherapy of the sacral area and has remained tumor-free seven (7) months since the operation.
Grossly, chordomas present typically as a lobulated tumor ranging from 5 to 15 cm in size. In most cases it is associated with extension beyond the bone and into the surrounding soft tissues.[5] Among the three (3) tumor variants, the dedifferentiated chordoma accounts for less than 5% of all chordomas.[4]
Patients’ symptoms largely depend on location but are most commonly neurologic. Pain is the usual initial presenting symptom. Obstructive symptoms, such as constipation, may also occur.[6]
Radiographically, chordomas are commonly located centrally, are solitary and lytic, and contain large areas of geographic destruction of tissues with scattered calcified areas. The tumors are hypo or isointense with T1 MRI, whereas they are hyperintense with T2 MRI.[6] Imaging cannot distinguish between the various subtypes.[4]
Microscopically, the presence of physaliphorous cells characterizes a chordoma. Physaliphorous cells have abundant pale, vacuolated or bubbly cytoplasm with small, hyperchromatic nuclei. These cells are admixed in a myxoid to chondromyxoid matrix and the tumor is divided into lobules by fibrous septa.1 Immunohistochemistry and other special techniques are not routinely required for arriving at the diagnosis in the presence of these associated histomorphologic features.[7] If these classic features are variably present, however, immunohistochemistry is warranted to rule out the closest histologic mimickers. Among the closest differentials are chondrosarcoma and metastatic renal cell carcinoma. Ruling out these differentials is important as they differ significantly from chordoma in terms of management and prognosis.
Positivity for CK, EMA, and S100 are traditionally used to support the diagnosis of a chordoma.8 This panel is generally sufficient to rule out the closest differential diagnoses (Table 1).

Table 1. Useful immunohistochemistry markers in ruling out differential diagnoses for chordoma
Immunoreactivity for these markers may be lost in the dedifferentiated areas. Should these prove insufficient, testing for Brachyury may be performed. It is the most sensitive and specific marker for tumors of notochordal origin, and it is completely absent in non-notochordal tumors. A study by Jambhekar et al., (2010) showed that Brachyury stained 90% of chordomas.
The mainstay of treatment for chordomas is surgical excision. Radiotherapy may be used in tumors near the skull base, and chemotherapy currently has no widely-accepted role.[9] Molecularly-targeted therapy has shown to be promising. A study by Casali et al., (2004) used the tyrosine-kinase inhibitor Imatinib mesylate for managing chordomas post-operatively.[10] The median disease-free interval was 32 weeks. This, among other treatments, are still under evaluation.
Tumor recurrence is high, generally associated with inadequate resection. The five (5)-year recurrence rate is 48%, while the 10-year rate is 67%.[1] The five (5) and 10-year survival rates are 45-77% and 28-50%, respectively. Metastatic disease is found in up to 43%[6] of cases either at the time of presentation or after excision.
Dedifferentiated chordoma is a rare and aggressive primary bone tumor. Surgery is the mainstay of treatment. No adjuvant treatment modalities have so far proven effective. It has high rates of recurrence and metastasis and carries an overall poor prognosis.
All efforts to secure patient's consent have been exhausted. The patient's anonymity is ensured. No other identifiers were included.
All authors certified fulfillment of ICMJE authorship criteria.
The authors declared no conflict of interest.
[1] WHO. Classification of tumours of soft tissue and bone, 4th edition. Lyon, France: International Agency for Research on Cancer, 2013.
[2] Hanna SA, Tirabosco R, Amin A, et al. Dedifferentiated chordoma: a report of four cases arising 'de novo.' J Bone Joint Surg Br. 2008;90(5):652-6. PubMed CrossRef.
[3] Meis JM, Raymond AK, Evans HL, Charles RE, Giraldo AA. “Dedifferentiated” chordoma: a clinicopathologic and immunohistochemical study of three cases. Am J Surg Pathol. 1987;11(7):516-25. PubMed.
[4] Chou WC, Hung YS, Lu CH, Yeh KY, Sheu S, Liaw CC. De novo dedifferentiated chordoma of the sacrum: a case report and review of the literature. Chang Gung Med J. 2009;32(3):330-5. PubMed.
[5] Fletcher C. Diagnostic histopathology of tumors, 4th edition. Philadelphia, PA: Saunders, Elsevier Inc., 2013.
[6] Rosai J. Rosai and Ackerman’s surgical pathology, 10th edition. USA: Mosby, Elsevier Inc., 2011.
[7] Dabbs DJ. Diagnostic immunohistochemistry, 4th edition. Philadelphia, PA: Saunders, Elsevier Inc., 2014.
[8] Jambhekar NA, Rekhi B, Thorat K, Dikshit R, Agrawal M, Puri A. Revisiting chordoma with brachyury, a "new age" marker analysis of a validation study on 51 cases. Arch Pathol Lab Med. 2010;134(8):1181-7. PubMed CrossRef.
[9] Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12(11):1344-50.
[10] Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer. 2004;101(9):2086-97. PubMed CrossRef.